

所述stetter反应是在所使用的反应有机化学,以形成碳-碳键通过1,4-加成利用反应的亲核 催化剂。[1]虽然相关的1,2-加成反应,苯偶姻缩合,自19世纪30年代以来就已知,但Hermann Stetter博士直到1973年才报道Stetter反应。[2]该反应提供了合成上有用的1,4-二羰基化合物和来自醛和迈克尔受体的相关衍生物。与1,3-二羰基不同,1,3-二羰基易通过Claisen缩合进入或1,5-二羰基,它使用的是通常由迈克尔反应,1,4-二羰基是具有挑战性的底物合成,但有若干有机转化,包括有价值的起始材料帕尔-克诺尔合成的呋喃和吡咯。传统上用于Stetter反应的催化剂是噻唑鎓盐和氰化物阴离子,但是最近对不对称Stetter反应的研究发现三唑鎓盐是有效的。Stetter反应是umpolung化学的一个例子,因为通过向醛中加入催化剂可以逆转醛的固有极性,使碳中心具有亲核性而不是亲电性。

机制
由于stetter反应是一个例子极性转换化学,所述醛是从转换电体为亲核反应条件下进行。[3]这是通过一些催化剂 – 氰化物(CN –)或噻唑盐的活化来实现的。[1]对于任何一种催化剂的使用,其机理非常相似; 唯一的区别是,对于噻唑鎓盐,催化剂必须首先去质子化以形成活性催化物质。活性催化剂可以描述为两种有贡献的共振形式的组合- 内鎓盐或卡宾两者都描绘了碳的亲核特征。噻唑鎓叶立德或CN –然后可以添加到醛底物,形成CN的情况下的羟腈–在噻唑鎓盐的情况下或布瑞斯罗夫中间。Breslow中间体由Ronald Breslow于1958年提出,并且是所有硫胺素催化反应的常见中间体,无论是体外还是体内。[4]

一旦形成“亲核醛” 合成子,无论是作为氰醇还是通过噻唑鎓叶立德稳定,反应都可以沿着两个途径进行。更快的途径是与另一种醛分子自缩合,得到安息香产物。然而,苯偶姻缩合是完全可逆的,因此不会干扰Stetter反应中的产物形成。实际上,可以使用苯偶姻代替醛作为底物以实现相同的整体Stetter转化,因为苯偶姻可以在反应条件下恢复其醛前体。[1]朝向Stetter产物的所需途径是将亲核醛1,4-加成到迈克尔型受体上。1,4-加成后,将反应是不可逆的,并最终,当催化剂踢出再生CN形成1,4-二羰基–或噻唑鎓叶立德。

范围
Stetter反应通常难以获得1,4-二羰基化合物和相关衍生物。传统的Stetter反应非常通用,适用于各种基材。[1]芳香醛,杂芳香醛和苯偶姻均可用作噻唑鎓盐和氰化物催化剂的酰基阴离子前体。然而,只有当噻唑鎓盐用作催化剂时才能使用脂族醛,因为当使用氰化物催化剂时它们经历醛醇缩合副反应。此外,α,β-不饱和酯,酮,腈,亚硝基和醛类都是合适的迈克尔受体和任一种催化剂。但是,不对称Stetter反应的一般范围更有限。分子内不对称Stetter反应基本上以任何组合享受一系列可接受的迈克尔受体和酰基阴离子前体。[5]分子内不对称Stetter反应可以利用芳香族,杂芳香族和脂肪族醛与拴系的α,β-不饱和酯,酮,硫酯,丙二酸,腈或Weinreb酰胺。已经表明,α,β-不饱和亚硝基和醛不是合适的迈克尔受体,并且在这种反应中具有显着降低的对映体过量。[5]分子内不对称Stetter反应遇到的另一个限制是只有导致六元环形成的底物才显示出合成有用的对映体过量; 形成五元和七元环的底物或者不反应或显示低立体诱导。[5]另一方面,分子间不对称反应非常局限于酰基阴离子前体和迈克尔受体的特别匹配的组合,例如脂族醛与硝基烯烃。[6]此外,这些底物倾向于相当活化,因为分子间不对称Stetter反应仍处于发展的早期阶段。

变化
自1973年发现Stetter反应以来,已经开发了几种变体。在2001年,Murry 等人报道了芳香醛与酰基亚胺衍生物的Stetter反应,得到α-酰胺酮产物。[7]酰基亚胺受体由α-甲苯磺酰胺底物原位产生,其在碱存在下经历消除。观察到良好至极好的产率(75-90%)。机械研究表明,与传统的Stetter反应相反,相应的苯偶姻不是足够的底物。[1]由此,作者得出结论,酰基亚胺的斯特特反应是在动力学控制下,而不是热力学控制。

Stetter反应的另一种变化涉及使用1,2-二羰基作为酰基阴离子中间体的前体。2005年,Scheidt及其同事报告使用丙酮酸钠,其失去CO 2以形成Breslow中间体。[8]同样,2011年,Bortolini及其同事证明了使用α-二酮生成酰基阴离子。[9]根据他们发展了的条件下,2,3- butadienone在加入到噻唑鎓催化剂释放乙酸乙酯并生成中间布瑞斯罗夫必需的stetter反应继续进行后切割。

此外,它们显示了原子经济性和使用环状α-二酮生成具有拴系乙酯的Stetter产物的效用。该反应通过与非环状形式相同的机制进行,但由乙醇侵蚀产生的酯仍然与产物连接。然而,由于需要乙醇作为溶剂,该条件仅允许产生乙酯。用叔丁醇代替乙醇不产生产物。作者推测这是由于两种醇溶剂之间酸度的差异。
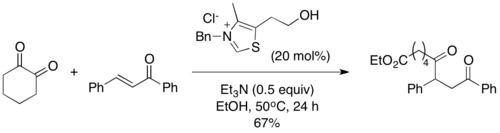
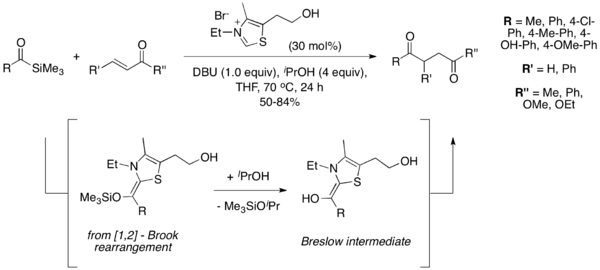
2004年,Scheidt及其同事引入了酰基硅烷作为Stetter反应中的主要底物,这种变化被称为“sila-Stetter反应”。[10]在它们的反应条件下,噻唑鎓催化剂诱导[1,2] 布鲁克重排,然后用异丙醇添加剂进行脱甲硅基化,得到传统斯特雷特反应的常见Breslow中间体。发现脱甲硅烷基化步骤是必要的,并且如果没有醇添加剂,反应不会进行。酰基硅烷比相应的醛具有更低的亲电子性,从而防止在Stetter反应中经常观察到的典型的苯偶姻型副产物。[11]
不对称的Stetter反应
Enders 等人于1996年报道了Stetter反应的第一个不对称变体,采用手性三唑鎓催化剂1。[12] 随后,报道了几种其他催化剂的不对称Stetter反应,包括2,[13] 3,[14]和4。[15]

Rovis集团的催化剂2的成功促使他们进一步探索这一系列催化剂并扩展其用于不对称Stetter反应的用途。2004年,他们报道了在略微改性的催化剂的分子内Stetter反应中,芳香醛的对映选择性形成季铵中心。[16]进一步的工作扩大了该反应的范围,包括脂肪醛。[17]随后,显示迈克尔受体的烯烃几何形状决定了这些反应中的非对映选择性,由此催化剂决定了初始碳键形成和烯丙基应变的对映选择性。最小化决定了非对映选择性分子内质子化。[18]

控制分子间反应中对映选择性的固有困难使得分子间不对称Stetter反应的发展成为挑战。尽管Enders在20世纪90年代早期报道了正丁醛与查尔酮的反应,但有限的对映体过量,[19]直到2008年恩德斯和罗维斯两组都发表这样的情况时才报道了合成有用的不对称分子间Stetter反应的条件。反应。Enders集团利用三唑鎓基催化剂实现芳香醛与查尔酮衍生物的偶联,产率适中。[20]来自Rovis集团的同时出版物也采用了三唑基催化剂,并报道了乙二酰胺和亚烷基丙二酸酯之间的Stetter反应,产率良好至极佳。[21]

Rovis及其同事随后继续研究杂环醛和硝基烯烃的不对称分子间Stetter反应。[22]在该反应过程的优化,我们发现具有氟化骨架的催化剂在反应中大大增强的对映选择性。有人提出氟化骨架有助于以增加对映选择性的方式锁定催化剂的构象。对该系统的进一步计算研究验证了立体电子在过渡态硝基烯上发展的部分负电荷与CF偶极子的部分正电荷之间的吸引力是使用具有主链氟化的催化剂观察到的对映体过量增加的原因。[23]虽然这是在分子间非对称stetter反应的区域中的显着进步,该基板范围是有限的,该催化剂为所用的特定底物进行了优化。

2011年,Glorius及其同事对不对称分子间Stetter反应的发展做出了另一个贡献。[6]他们通过利用N-酰胺基丙烯酸酯作为共轭受体,证明了α-对氨基酸的对映选择性合成。值得注意的是,反应可以在5mmol规模下进行,而不损失产率或对映选择性。

应用
Stetter反应是有机合成中的有效工具。Stetter反应的产物1,4-二羰基化合物是用于合成复杂分子的有价值的部分。例如,Trost及其同事采用Stetter反应作为合成rac- hirsutic acid C的一步。[24]脂肪醛与拴系的α,β-不饱和酯的分子内偶联导致所需的三环1,4 – 二羰基,产率67%。将该中间体再七步转化为rac- hirsutic acid C.

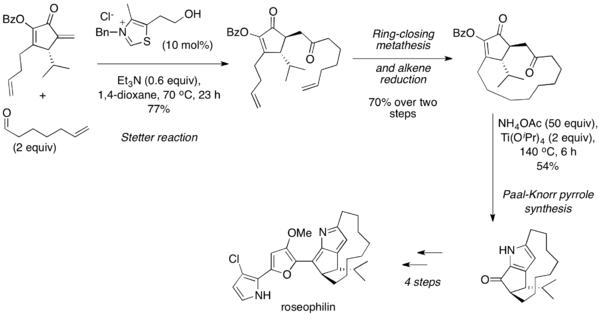
Stetter反应通常与呋喃和吡咯的Paal-Knorr合成顺序使用,1,4-二羰基在高温酸性条件下与其自身或在胺存在下进行缩合。2001年,Tius及其同事报道了利用分子间Stetter反应将脂肪醛与环烯酮偶合的玫瑰花青素的不对称全合成。[25]之后闭环复分解和烯烃还原,1,4-二羰基的产物经由帕尔-克诺尔合成转化为吡咯和进一步阐述的天然产物。
2004年,报道了一锅偶联 – 异构化 – Stetter-Paal Knorr序列。[26]该过程是先利用钯交叉偶联化学与炔丙醇耦合的芳基卤化物,得到α,β不饱和酮,其然后可以经历与醛stetter反应。一旦形成1,4-二羰基化合物,在酸存在下加热将得到呋喃,而在氯化铵和酸存在下加热将得到吡咯。整个序列在一锅中进行,步骤之间无需后处理或纯化。

Ma和同事开发了一种利用Stetter反应获取呋喃的替代方法。[27]在他们的报告中,在Stetter条件下合成3-氨基呋喃以使芳香醛与乙炔二羧酸二甲酯(DMAD)偶联,由此噻唑鎓叶立德通过呋喃产物的芳构化而水解。由于噻唑鎓在这些条件下被破坏,它不是催化剂,必须以化学计量的量使用。
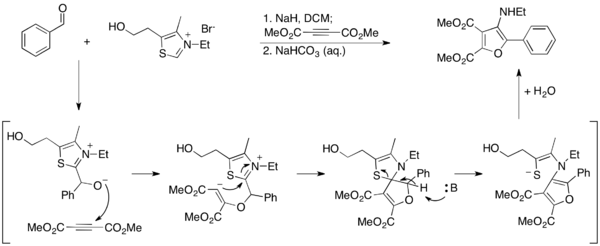
他们通过开发一种方法进一步阐述了这项工作,其中2-氨基呋喃通过环化合成腈来合成。[28]在这种方法中,噻唑鎓叶立德催化采用并产生游离胺产物。





