

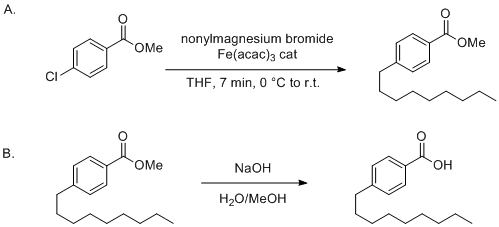
4-壬基苯甲酸
 |
讨论
在很大程度上受生命科学和材料化学需求的驱动,对更加有效的交叉耦合程序的需求似乎并未停止。虽然基于钯的方法继续在该领域占据主导地位,但过去十年见证了许多创新概念的发展,这些概念丰富了从业者的投资组合,甚至以某种方式挑战有机钯催化。
其中一个发展涉及使用早期过渡金属代替钯(或其他贵金属); 实际上,这个大趋势远远超出了交叉耦合化学。2功利主义论证使铁成为一种特别有吸引力的候选者,因为它价格便宜,丰富,容易获得,对环境无害且对人类几乎没有毒性。此外,对均相有机铁催化的研究有望通过在元素周期表中利用该元素的独特化学性质来开辟全新的应用领域。2,3
关于使用铁催化剂进行“交叉耦合”的分散报告可以追溯到实际创造这种表达之前的时间。第一故意调查报告了高知和同事,4,5谁表明,简单的铁盐是能够催化格氏试剂的耦合与链烯基卤化物。由于收益率可变且反应似乎缺乏一般性,因此只有少数应用程序跟进此主要发现。Cahiez及其同事发现,通过使用非质子偶极共溶剂,优选N-甲基吡咯烷-2-酮(NMP),可以大大提高该过程的稳健性,这似乎可以稳定实际的催化剂和/或活化瞬时形成的有机铁中间体。 。6,7
规范交叉耦合
我们的研究小组表明,底物范围远远超出了烯基卤化物。2002年,我们报道了芳基 – 和杂芳基卤化物和 – 磺酸盐的第一次铁催化交叉偶联反应(图1)。8,9,10,11,12之后不久,烯基三氟甲磺酸酯(图2)和酰基氯被认为是同样特权偶联伴侣。132005年有机合成程序描述了4-壬基苯甲酸的制备液晶材料的一个组成部分,很好地捕捉了这种铁催化剂的发展状态,因为它突出了以下显着的优点:(i)带有β-氢原子的格氏试剂的高产率,(ii)特别快的反应速率(或室温下,(iii)准备好的可扩展性,(iv)方便的无配体条件,和(v)相对于许多对Grignard试剂的非催化攻击敏感的官能团,具有令人惊讶的大的耐受性。同样重要的是,与相应的溴化物或碘化物相比,缺电子(杂)芳基氯化物被证明更适合于铁催化的交叉偶联。然而,当使用富电子(杂)芳烃时,必须使用相应的三氟甲磺酸酯。8,9,11此外,有可能以接合运载多于一种氯化物或磺酸盐基团的基材或者成穷举,或位点选择性或连续一锅偶联反应。9, 10, 11, 12, 13
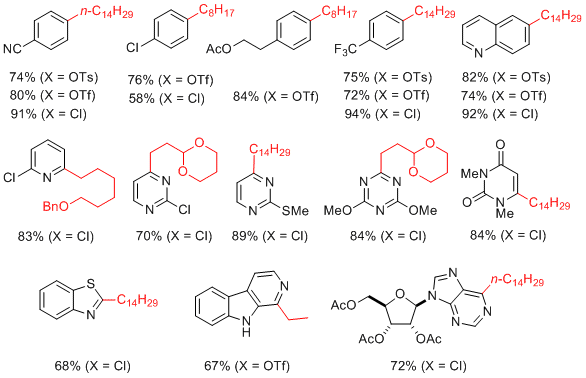
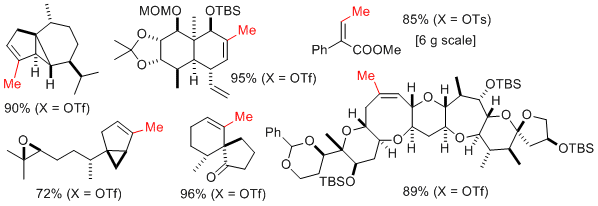
图1.铁催化的烷基/(杂)芳基交叉偶联反应; 9(在整篇文章中,来自Grignard试剂的片段显示为红色)
图2.铁-催化的链烯磺酸盐的交叉偶联用MeMgX 13,27,28,29,30,31
鉴于这些有利的属性,学术和工业界迅速接受这种化学反应可能并不令人感到意外。14,15,16,17,18在图3所示的选择的更大规模的应用是有启发性和示出了现有技术的当前状态。19,20,21,22,23,24,25,26
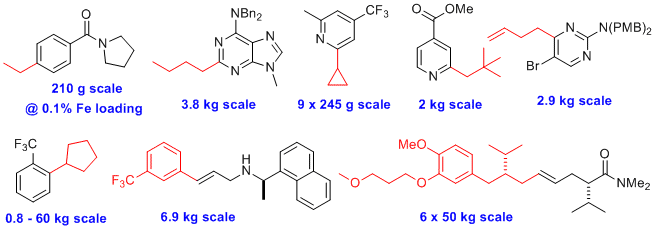
图3.应用在较大规模19,20,21,22,23,24,25,26
我们的初步报告已经表明,使用Fe(acac)3作为简单而有效的预催化剂,芳基三氟甲磺酸酯和甚至(缺电子的)芳基甲苯磺酸酯可与烷基 – 格氏试剂交叉偶联,产率良好至极佳(参见图1) 。8,9个的链烯基磺酸盐此后不久,接着(图2); 13,27,28最近的组织。合成。程序进一步说明了其效用 29在这些引线发现大厦,反应的范围进行了探讨30,31,32,33及其他可行的离去基团进行了鉴定,包括膦酸盐,23,32,34,35,36,37个氨基磺酸盐,32,38,39点的氨基甲酸酯,38,62个羧酸盐,40,64三烷基铵盐,65和甚至各种(硫代)醚。61,63的几个选择的实施例列于表1中。
虽然铁盐的成本几乎不是限制因素,但是试图降低负荷; 使用≤1mol%的应用并不少见。20,41,42不吸湿,因此实际的Fe(ACAC)3是最流行的预催化剂; 在某些情况下,其它的盐如的FeCl Ñ(Ñ = 2,3),FEF 3 -3H 2 O,38,49或铁硫醇盐43表现出更好的性能。同样,许多铁络合物(非原位或原位形成)显示出良好的应用特征。
NMP仍然是铁催化的交叉偶联反应的优选共溶剂(尽管某些反应在其不存在时效果更好)。然而,该化合物具有潜在的毒性。因此,进行了许多尝试以完全避免使用它或用更良性的添加剂,溶剂或配体代替NMP。6,29,44,45,54其中,环状脲,如N,N-二甲基咪唑啉-2-酮(DMI)55和各种(二)胺,特别是TMEDA(N,N,N”,N’-四甲基乙二胺),29,46,47,48最常见的; 然而,它们的使用通常要求将Grignard试剂缓慢加入反应混合物中。此外,许多报道依赖于使用NHC或(螯合)(二)膦。49,50,51,52,53
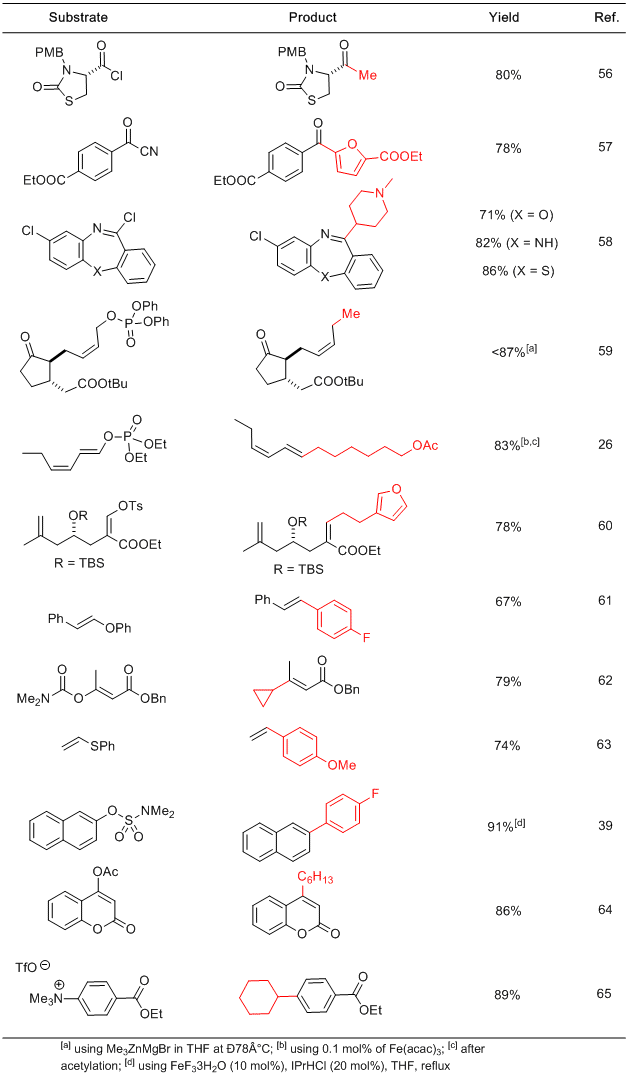
表1.铁催化交叉偶联:代表性实例
尽管有这些进步,但也需要提到一些重要的限制。与钯催化的交叉偶联反应相反,后者在形成C(sp 2)-C(sp 2)或C(sp 2)-C(sp)键时表现优异,最显着的是芳基 – 芳基键,对于这个目的,铁催化目前还不够; 竞争同源偶联格氏试剂限制了范围。尽管已经报道了进展,但是在这个特别重要的应用领域中真正可与钯(或镍)催化剂相媲美的通用解决方案目前仍然是难以捉摸的。9,38,39,49,66,67,68, 75
Grignard试剂是特权亲核试剂的事实同时表明了优势和严重的限制。它们便宜且在工业上可行,但在功能组兼容性方面明显受限。然而,许多铁催化的交叉偶联反应的异常速率部分地减轻了这个缺点:因此,已经成功地使用了许多底物,其带有亲电取代基,否则在有机镁试剂存在下可能不存在亲电取代基。该名单包括:烷基 – 和芳基卤化物,酰胺,氨基甲酸酯,烯酸酯,环氧化物,酯,异氰酸酯,酮,腈,磺酸酯,磺酰胺,硫代氨基甲酸酯; 然而,这种动力学选择性可能与情况有关。因此,需要继续扩大适当亲核试剂的组合。69, 70 -锌, 59, 71, 72, 73 -锂, 74 -铜, 75种-硼试剂。76, 77, 78, 79, 80
在本文中,要指出的是,烷基/芳基交叉偶联也可以在不单独步骤中制备格氏试剂的情况下进行。81,82作为一个例子,处理环戊基溴和的混合物的1-溴-2-(三氟甲基)苯与镁和催化量的FeCl 2在环境温度下在THF / TMEDA,得到1-环戊基-2-(三氟甲基)苯的产率为67%,最高可达60公斤(图3)。83由于瞬时形成的格氏试剂直接消耗,因此在任何时间点都不会积聚大量浓度。
非规范交叉耦合
廉价和良性铁催化剂允许钯取代的前景 – 至少在某些交叉偶联反应中 – 是(并且是)主要的诱因。6,8,9它很快变得明确,但是,认为这是金属也能促进具有在元典很少或没有先例基板的交叉偶联反应。
初级和次级烷基卤化物参与与芳基卤化镁和其他亲核试剂的铁催化交叉偶联的准备就绪说明了这一点。47,48,50,51,84,85,86,87,88,89,90,91官能团耐受性显着,甚至空间位阻的烷基卤通常反应良好(表2)。此外,探索性研究表明,外消旋仲烷基卤化物的不对称对映聚合交叉偶联可能与带有螯合二膦配体的铁催化剂在磷上是手性的(方案1)。92
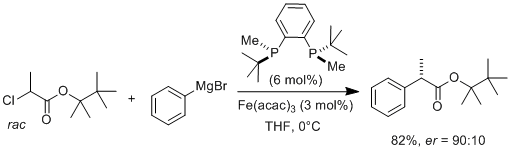
方案1.不对称铁催化的芳基/烷基交叉偶联的开创性研究92
即使不太常规的基板也能很好地工作(表2)。例如,1-炔基环丙基甲苯磺酸酯在Fe(acac)3 cat 存在下与格氏试剂反应。净炔基取代; 112此引人注目的结果说明,即使叔烷基电体可以成功地从事铁-催化的交叉偶联。52,112然而,这个图案对比大多数其他炔化合物,其通常用S得到丙二烯的行为Ñ 2’型取代反应(除一些炔丙基溴化物)。93,94,95,96,97,113对于炔丙基环氧化物,显示RMgX试剂接近O-原子的顺式,而有机铜试剂将R-取代基反式递送至离去基团。95,113
非常规的交叉偶联反应在内酯衍生的宝石 – 二氯烯烃转化为非末端炔烃中表现出来(方案2)。98,99,100。注意,有机锂试剂的R基团被结合到产品作为取代基封端的炔; 这种转化可能通过类胡萝卜素中间体进行。甚至更多涉及的交叉耦合“级联”在文献中已知(方案2)。101,102
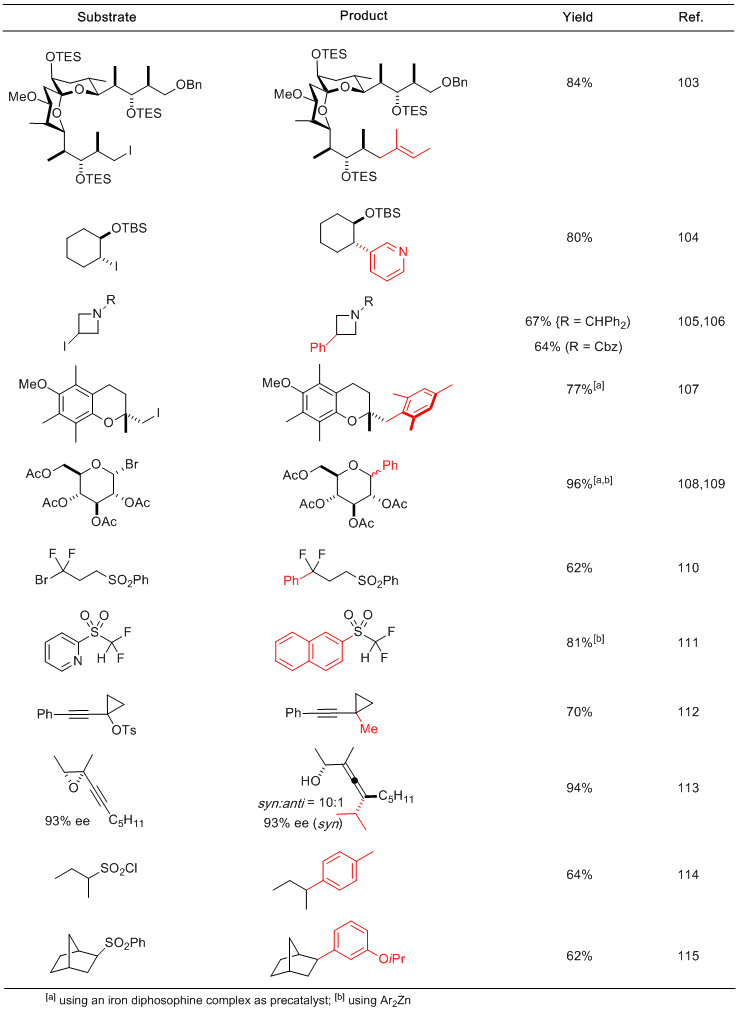
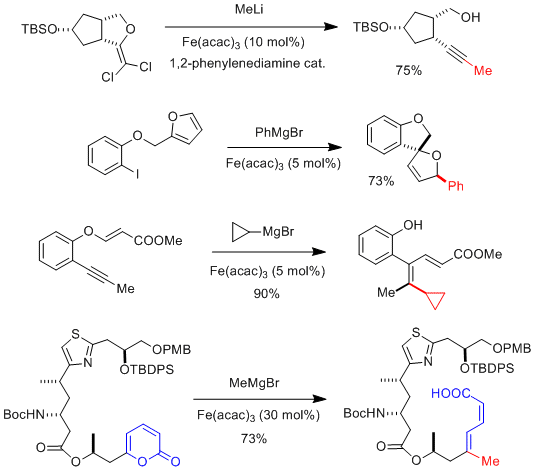
表2.非正统亲电子试剂的铁催化交叉偶联
方案2.某些非正统的铁-催化的“交叉偶联”反应99,101,102,118
2-吡喃酮衍生物的铁催化的开环/交叉偶联是相当独特的。116在形式上,嵌入杂环中的烯醇酯部分用作离去基团。然而,可用的机理证据表明,原位形成的铁催化剂不会插入CO键中; 更确切地说,与吡喃酮π-体系的配位被认为引发1,6-加成/开环级联。环开放步骤本身是电环过程还是遵循离子机制仍然是牢固建立的。无论如何,这种化学物质与官能团和各种供体位点相容; 它可以获得非热力学二烯酸酯,否则难以以异构纯的形式制备。在有效的抗癌剂pateamine A的全合成期间的后期应用说明了这一方面(方案2)。117,118
本讨论附录不能提供广泛定义的铁催化交叉偶联的综合论述; 14,15,16,17,18,而,所选择的实施例意在举例说明,因为原来在该领域的巨大进步组织。合成。该程序于2005年出版。虽然可以安全地预测进一步的增长,但是铁催化作用一直在等待更好的机械理解。2大规模的分析挑战尽管如此,近期取得实质性进展提供了一个令人鼓舞的前景。




