

的炔三聚反应是2 + 2 + 2环化反应,其中三个分子的炔反应形成芳烃。该反应需要金属催化剂。该过程具有历史意义,也适用于有机合成。[1] 已经开发了许多变体,包括炔烃和烯烃以及炔烃和腈的混合物的环化。
历史
1948年,Reppe发现镍化合物催化乙炔形成取代的苯:[2] [3]
- 3 RC 2 H→C 6 R 3 H 3
观察到1,3,5-和1,2,4-C 6 R 3 H 3异构体。
自从Reppe的发现以来,已经报道了许多其他的cyclotrimerizations。[4] [5] [6] [7] [8] [9]
机制和立体化学
没有金属催化作用,反应就不会发生。反应开始于金属 – 炔烃配合物的形成。配位球内的两种炔烃的组合提供金属环戊二烯。[10]从金属环戊二烯中间体开始,通常讨论两种途径:
- (1)第三炔插入产生金属环庚三烯。 直接从金属环庚三烯中还原消除形成芳烃是对称性的,[11]但已经证明了逐步机制。
- (2)第三炔烃参与[4 + 2]环加成反应,生成双环物质(金属降冰片二烯)。
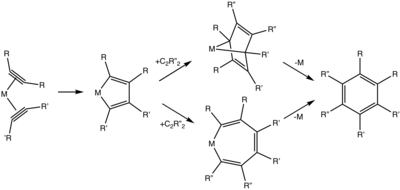
炔三聚的两种途径。Coligands被省略。[10]
对于不对称取代的乙炔的环三聚反应,关于产物芳烃的取代模式分两步测定:金属环戊二烯中间体的形成和第三当量炔的掺入。虽然头对头和尾对尾金属环戊二烯均为1,但许多乙炔基质选择性地形成2型区域异构体。已经引用炔烃偶联配偶体和催化剂上的空间体积作为区域选择性的控制元素。[12]
手性催化剂已与芳烃组合使用以产生非外消旋的阻转异构产物。[13]
范围和限制
用于环三聚的催化剂对三键具有高选择性,这使反应具有相当宽的底物范围; 醇类,醚类,胺类和羰基化合物(酮类,酯类,酰胺类和羧酸类)都具有良好的耐受性。[ 引证需要 ] 腈类可以与炔烃反应生成吡啶。[14]
另外,一些反应通过催化剂失活通过形成稳定的,18-电子η的限制4个 -complexes。[15] 环丁二烯,环己二烯,和芳烃络合物都被观察为关断周期,失活催化剂的形式。除了源自低区域和化学选择性的高级聚合物和二聚体和三聚体之外,还观察到衍生自炔二聚的烯炔副产物。铑催化剂特别适用于烯炔形成(见下文)。[16]对于镍催化,形成较大的环(特别是环辛四烯)可能是个问题。

综合应用
用单独的炔烃环化二炔可以提供具有高原子经济性的稠环系统。在合成雌酮的一个引人注目的例子中,下面所示的二炔反应物与二(三甲基甲硅烷基)乙炔结合产生苯并环丁烯产物[17]。加热后,开环产生醌甲基化物,参与随后的分子内Diels-桤木反应。

如果第三炔单元被束缚于所述第一两个,三个环可在单一步骤中被创建。在下面合成calomelanolactone的例子中,Wilkinson催化剂用于催化所示三炔的分子内共聚。[18]

拥挤的三炔可以环化成具有螺旋手性的产品。在一个实施例中,对于在一个步骤中形成三个新芳环而言显着,通过用环戊二烯基钴二羰基处理将所示的三炔转化为螺旋产物。[19]截至2004年,这个过程尚未变得不对称,[ 原创研究?]但产品可通过手性HPLC分离。[19]

与其他方法比较
环三聚反应提供了通过亲电或亲核取代使预形成的芳族化合物官能化的替代方案,其区域选择性有时难以控制。
从取代的不饱和前体直接形成芳环的其他方法包括Dötz反应,钯催化的烯烃与炔烃的[4 + 2] 苯并环化,[20]和路易斯酸介导的[4 + 2]环烯烃环加成反应与炔烃。[21]钯催化的瞬时苄基物种与炔烃的环化也可以产生取代的芳族化合物。[22]





